File:US System of Oversight of Genetic Testing p242 C-4 Research and Development - 2008 SAGCHS.png

{kind=link}
Original file (990 × 1,333 pixels, file size: 252 KB, MIME type: image/png)
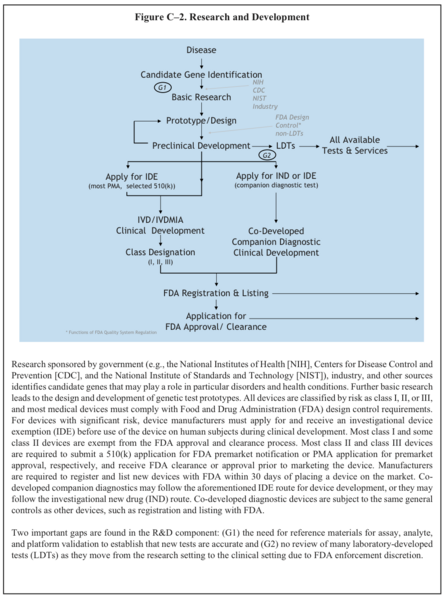
"Research sponsored by government (e.g., the National Institutes of Health [NIH], Centers for Disease Control and Prevention [CDC], and the National Institute of Standards and Technology [NIST]), industry, and other sources identifies candidate genes that may play a role in particular disorders and health conditions. Further basic research leads to the design and development of genetic test prototypes. All devices are classified by risk as class I, II, or III, and most medical devices must comply with Food and Drug Administration (FDA) design control requirements. For devices with significant risk, device manufacturers must apply for and receive an investigational device exemption (IDE) before use of the device on human subjects during clinical development. Most class I and some class II devices are exempt from the FDA approval and clearance process. Most class II and class III devices are required to submit a 510(k) application for FDA premarket notification or PMA application for premarket approval, respectively, and receive FDA clearance or approval prior to marketing the device. Manufacturers are required to register and list new devices with FDA within 30 days of placing a device on the market. Co- developed companion diagnostics may follow the aforementioned IDE route for device development, or they may follow the investigational new drug (IND) route. Co-developed diagnostic devices are subject to the same general controls as other devices, such as registration and listing with FDA.
Two important gaps are found in the R&D component: (G1) the need for reference materials for assay, analyte, and platform validation to establish that new tests are accurate and (G2) no review of many laboratory-developed tests (LDTs) as they move from the research setting to the clinical setting due to FDA enforcement discretion."
File history
Click on a date/time to view the file as it appeared at that time.
| Date/Time | Thumbnail | Dimensions | User | Comment | |
|---|---|---|---|---|---|
| current | 09:51, 2 October 2009 | | 990 × 1,333 (252 KB) | Mac (talk | contribs) | [[Diagnostic_Kits/Secretary's_Advisory_Committee_on_Genetics|U.S. System of Oversight of Genetic Testing: A Response to the Charge of the Secretary of Health and Human Services, Report of the Secretary’s Advisory Committee on Genetics, Health, and So |
You cannot overwrite this file.
File usage
The following 2 pages use this file:
{kind=link}